For a seemingly simple question, the answer yields enough content to be covered over the course of an entire semester (maybe even two). Don’t worry; you did not accidentally enroll in Professor Ben’s Pharmacokinetics 101 class; there will be no final exam or mid-year review. I know what you’re thinking, how will I try to cover a semester’s worth of content in one blog post? My answer is simple. I won’t, but I will focus on a simplified view of systemic pharmacokinetics to help cultivate the foundation that we can later build on through supplemental blog posts.
First things first, let’s break down the root of the word to its simplest form. Pharmaco- is a Greek prefix that means “drug,” and -kinetics is a Greek suffix that means “in motion.” So essentially, Pharmacokinetics is the study of “Drugs in Motion.” In my Introduction post, I described why these processes are so fascinating to me (if you haven’t checked out that post yet, I highly recommend it).
Typically, when the PK of an active pharmaceutical ingredient (API) is described, it is the plasma PK rather than tissue PK. Why does this matter? The plasma API concentrations are assumed to be in equilibrium with the surrounding tissues. Therefore, these plasma concentrations are surrogates for the tissue concentrations, or at the site of action(s), due to this assumed dynamic equilibrium. This is a fairly complex notion yet might be problematic. Briefly, if the plasma concentrations are above the minimum effective concentration, this may indicate that the API concentration is sufficient to elicit the therapeutic effect in the tissues. However, studies have suggested that plasma concentrations may not always be equivalent to tissue concentrations. [1] These subtherapeutic tissue concentrations may give rise to drug resistance over time, and thus, tissue concentrations are the “gold standard” still are not always practical, feasible, or ethical to obtain.
Now let’s dive into exactly what pharmacokinetics (PK) means and how to define the processes that occur within the body. What are the processes involved with PK? In its simplest form, the concentration-time profile can be described by 4 (or 5) different processes – ADME, which stands for absorption, distribution, metabolism, and elimination (or excretion). In any dosage form, the drug must first be liberated from the dosage form to be absorbed into the body, and thus you may see it written as LADME, where the “L” stands for liberation. I will briefly explain each of these processes that occur once a drug product is administered to a patient. For simplicity in describing these processes, we will use the API in a tablet form. (Topical formulations will be used in the upcoming cutaneous PK post 🙂 )
Liberation
The active pharmaceutical ingredient (API) must escape the drug product (tablet, capsule, cream, gel, etc.) to be available for absorption. This process can also be referred to as a dissolution process, which describes the API’s rate to be absorbed; it is also the rate of escaping the drug product. In this case (Figure 1), the tablet contains not only the API but also other additives, which are termed excipients. These excipients are inactive ingredients with a range of abilities, from solubilizing the API to stabilizing the formulation. Once administered, the excipients and API begin to dissociate, allowing the API to be absorbed either in the duodenum/small intestine or the large intestine if the drug is acid labile. If the API is acid labile, the tablet will be enteric-coated to protect against degradation while in the stomach. The rate-limiting step in the PK of an API is often the absorption from the dosage form, emphasizing the absorption processes’ importance.
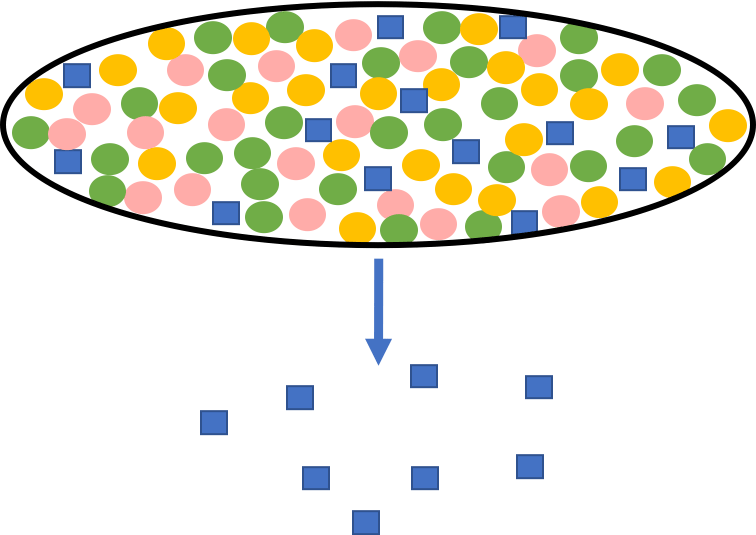
Absorption
Now that the API is free in solution, it can be absorbed into the systemic circulation. The first step is for the API to be absorbed as it traverses down the gastrointestinal tract, where the duodenum is often targeted for rapid absorption. After the API is carried to the liver, it will then go into the systemic circulation. It is important to note that there is no absorption step when delivering therapeutics directly into the blood through intravenous (IV) infusion or IV bolus administration. In conjunction with the clearance of a compound, the absorption plays an important role in the bioavailability (BA) of an API.
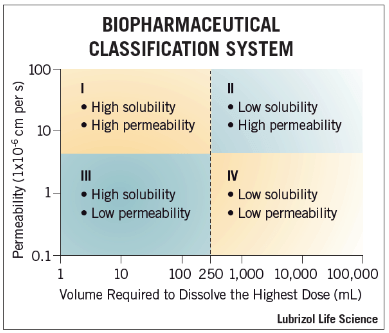
The BA is a fraction of how much API is available within the systemic circulation and available to go to the desired site of action to reach the minimum effective concentration. Another important concept (Figure 2) is the Biopharmaceutics Classification System (BCS) (Post to come – something that is also being worked on for topically applied drug products!) where the degree of solubility and permeability determines the classification of an API. While this classification system allows for a general understanding of how an API may behave in vivo, many factors impact the absorption of a compound, such as molecular weight, solubility, hydrogen bonds donors/acceptors, or octanol-water (Log-p) partition coefficient. These are known as the Lipinski Rule of 5, which may also apply to topical drug delivery. Once the API has been absorbed into the systemic circulation, the formulation no longer plays a role in the following processes. When looking at a concentration-time profile, there is a maximum concentration that is reached (Cmax) when the rate going into the sampling compartment (absorption rate) is equal to the rate going out of the compartment (elimination rate). The time at which Cmax occurs is termed the tmax. As the bioavailability typically depends on the absorption rate to the sampling compartment, Cmax becomes an important term that we will focus on in future posts.
Distribution
Distribution is the process by which the API permeates into various tissues from the systemic circulation. The degree of distribution depends on the tissue vascularization and the API’s ability to permeate across the blood vessels and cells. Therefore, APIs are typically highly distributed into highly perfused organs such as the heart, liver, and kidneys. Now you may say, “Wait a minute, Ben, you said highly perfused organs – why not the brain?” You would be right to question this, yet the brain has a complex defense system against non-native molecules to keep the thinking machine operating correctly. The brain is an example of permeability-limited distribution, as many enzymes/transporters will actively kick out APIs that cross the blood-brain barrier. Other tissues such as muscles or fat are often termed flow-limited as the API can permeate into the tissues; it is just a matter of getting to the tissues. A parameter used to describe the APIs distribution that we will investigate in subsequent posts is termed volume of distribution (Vd). As a human participant’s blood volume is approximately 5 liters, a Vd less than 5 indicates that the API stays in the blood. In contrast, a value larger than 5 indicates the API is extensively distributed throughout the tissues. Vd is nothing more than a proportionality constant to help relate the amount of API in the body to the plasma concentrations.
Another factor that influences the distribution of an API is protein binding. It is impossible to talk about Vd without talking about the protein binding or relative fraction unbound (funbound). “The total drug once in the body is available to go to the site of action, right, so who cares about the unbound drug, Ben?” Wrong, only the unbound drug is responsible for the pharmacological action. [2, 3] The total API will be analyzed via liquid chromatography, but the funbound is estimated before a preclinical or clinical study to relate the two. An API might bind plasma and tissue proteins alike, but the major source of API binding is albumin. However, these binding events are non-covalent and, as such, are reversible based upon the concentration gradient. Protein binding also acts as a depot for the API as there is an equilibrium between bound and unbound in the systemic circulation. As the more unbound drug is eliminated, the more bound drug then becomes unbound. This process iterates until there is no drug left in the system. The two scenarios that may affect the API’s disposition are a high or low protein-bound API. Consider an API that is 95% protein-bound, and due to drug-drug interaction, the protein binding is now 90%. We have a 5% increase in unbound API, but we have a 100% increase in unbound API circulating the system compared to the baseline binding. If we consider a low protein binding baseline of 10% and a drug-drug interaction increases the bound API to 15%, we now have roughly a 5% increase in unbound API but at a 50% increase in the amount circulating the system. “So a protein-binding change is the determining factor in systemic API exposure, right?” Maybe. Without going too in-depth (future post), if the API’s clearance is not blood flow-dependent, it doesn’t matter, but if the API’s clearance is blood flow-dependent, protein binding is of utmost importance. [4]
Metabolism
The concept of API clearance is made up of two sub-domains: metabolism and excretion. Metabolism is the breakdown of the parent API into more hydrophilic molecules such that the kidneys can eventually excrete the API. The major metabolic activity occurs within the liver, where lipophilic compounds are broken down to be excreted in the urine; Hydrophilic compounds are more easily excreted in the urine. The enzymes that breakdown the lipophilic molecules have subdivisions: Phase I and Phase II. Phase I processes include oxidation, reduction, and hydrolysis, while Phase II metabolism includes acetylation, glucuronidation, and sulfonation. It may also be that the molecule formulated is not active itself but once in the body; it can be rapidly metabolized and then activated, which is called a pro-drug. Another essential concept is the first-pass metabolism, which is a major hurdle that APIs must overcome to get into the systemic circulation. As I mentioned above, once the API is liberated from the formulation and is now is a solution within the body, the API will pass through the hepatic portal vein and then into the systemic circulation. If the API has a relatively small fraction absorbed after the first-pass metabolism, strategies must be devised to allow more input of API into the system and thus the site of action. One of the most important enzymes that breakdown compound is the cytochrome P450 (CYP-P450) family of enzymes. These are particularly important when we consider drug-drug interactions as many times a subject is on multiple therapeutics simultaneously. There is a comprehensive review of the substrates, inhibitors, or inducers for the various CYP enzymes – Flockhart TableTM.
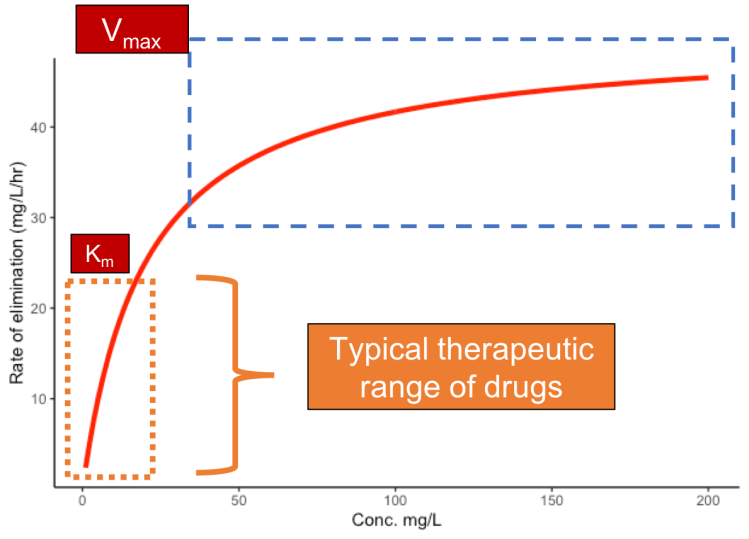
Disease states can alter these enzymes/transporters, and therapeutics may need to be adjusted to either reduce potential toxicity or increase efficacy. These alterations can be understood via Michaelis-Menten kinetics, which describes the linearity or nonlinearity by which a drug is metabolized. Michaelis-Menten kinetics are always occurring, but typically higher API concentrations lead to a nonlinear change in the resulting metabolic product. Depending on the API’s therapeutic dose, this might be important and is often evaluated in the ADME drug discovery process.
Excretion (Elimination)
The “E” in ADME is almost always excretion, so why do I have elimination in parentheses, you might ask. In tissue-specific PK, the API is eliminated via the blood and possibly lymphatic system. It will eventually be excreted via the kidney, but, in my opinion, this is why you might use elimination interchangeably, depending on the type of PK you are investigating. Once the API is more hydrophilic (or water-soluble), either native or via metabolism, it can then be excreted in the urine. Excretion occurs in the kidneys via glomerular filtration and active secretion in the peritubular capillaries, where filtration accounts for most API removal. Diseases affecting the kidneys may significantly increase bioavailability if an API is mainly removed via glomerular filtration. In addition to measuring the plasma concentrations, one may also quantify the amount of API excreted unchanged in the urine to comprehend the relative bioavailability and the fraction of the dose eliminated. To gain insight into which elimination process dominate, the total clearance can be compared to the relative excretion rate.
Wrapping It All Up
After reading this post, I hope that some of the overarching concepts in PK have been conveyed simplistically. There are plenty of locations where I indicated that I would be writing future posts, and I will update these with links to actual posts in the future! This is only the foundation for things I will be exploring/explaining, and eventually, I will begin to talk about cutaneous PK, but first, we need a solid understanding of systemic PK of these concepts and equations (oh no, I said equations – please come back :)) so please bear with me as I begin to dive deeper. As I may not have explained every single thing about PK so feel free to drop a comment below and/or come back for future blog posts!
Happy New Year – I hope that 2021 is the best year for everyone yet!
References
- Fischman, A.J., Alpert, N.M., Livni, E., Ray, S., Sinclair, I., Callahan, R.J., Correia, J.A., Webb, D., Strauss, H.W., and Rubin, R.H., 1993. Pharmacokinetics of 18F-labeled fluconazole in healthy human subjects by positron emission tomography. Antimicrobial agents and chemotherapy, 37(6), pp.1270-1277.
- D. Gonzalez, S. Schmidt, H. Derendorf, Importance of relating efficacy measures to unbound drug concentrations for anti-infective agents, Clin Microbiol Rev, 26 (2013) 274-288.
- E.C. de Lange, P.G. Ravenstijn, D. Groenendaal, T.J. van Steeg, Toward the prediction of CNS drug-effect profiles in physiological and pathological conditions using microdialysis and mechanism-based pharmacokinetic-pharmacodynamic modeling, The AAPS Journal, 7 (2005) E532-E543.
- Benet, L. Z., & Hoener, B. A. (2002). Changes in plasma protein binding have little clinical relevance. Clinical Pharmacology & Therapeutics, 71(3), 115-121.

One thought on “What is Pharmacokinetics?”